
 400-800-6815
400-800-6815
 返回列表
返回列表

CellPro 赛普应用与验证中心
拥有独立的PCR实验室、细胞培养实验室、微生物实验室
自动化工作站适配实验室等
能够进行一站式的耗材、试剂、仪器的
生物学性能验证与整合研发
今日CellPro 赛普实验课堂上线
给大家分享PCR实验技术及问题解决方案
样本采集和保存
良好的样品采集和保存方法对于DNA提取的成败至关重要,尤其对于珍贵样品的采集和保存的方式更是需要特别小心和关注。

动物组织样本:
l 大块组织样本,取材后切成小块,PBS清洗2-3次,保存于无菌管;
l 呼吸道样品通过棉拭子蘸取采集,完成采样后应立即置于磷酸盐缓冲液中;
l 血液样品采集后需要立即加入抗凝剂,竖立插入样品盒。
植物组织样本:
l 植物样品采集时优先选取细胞分裂旺盛,核酸含量较多的部位,如植物的幼嫩部位;
l 采集植物样品时建议使用无菌水或75%乙醇来擦拭冲洗,再用吸水纸吸干样品表面液体。
细胞样本:
l 离心收集沉淀后即可使用。


大部分情况下应尽可能使用新鲜制备的样品进行后续实验,以期获得最好的结果。但客观情况是刚采集的组织样品是不能马上进行DNA提取,短期内保存运输可以置于-20℃~0℃,长期保存可直接通过液氮速冻并置于-80℃保存半年。

DNA提取纯化

该方案是用酚类试剂变性蛋白质,来对样本进行裂解,后用苯酚/氯仿抽提,再无水乙醇沉淀。氯仿的作用是去除多余的酚,促进水相与有机相的分离。该提取方法需要多次离心,步骤较为繁琐,易造成交叉污染。另外,由于终产物中会引入苯酚/氯仿等有机溶剂残留,对后续PCR实验可能会有抑制。

是通过加入各种蛋白酶去除蛋白杂质,从而分离DNA,有效避免试剂的污染,以获得高产量和高纯度的DNA,但缺点是消解蛋白酶的过程费时较多,且纯度不稳定。

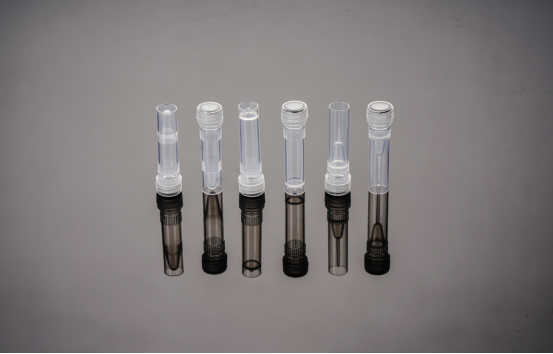
该方案是将 DNA 分子中的磷酸二酯骨架在离液盐作用下脱水后,使得磷酸基团暴露的同时与硅胶发生可逆性吸附。硅介质吸附纯化法可以通过离心以及真空加压的方法使裂解液穿过滤膜,可以有效的提取微量的 DNA。静电力和氢键在硅胶与核酸的吸附中起着关键作用。该方法在脱氧核糖核酸的链长上有一定限制,较小的 DNA 片段(<100 bp)不容易在介质上得到有效吸附。

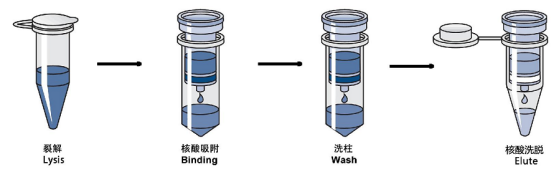
该方法将纯化介质包被在纳米级的磁珠表面,通过介质对DNA的吸附,在外加磁场的作用下使DNA附着于磁珠并定向移动,从而达到核酸与其它物质分离的目的。
与其它的方法相比,磁珠法有以下优势:
l 分离快:磁场分离只需几秒钟;
l 提取灵敏度高:微量的样本也可纯化分离;
l 纯化纯度高:核酸与杂质分离完全;
l 提取产量高:每毫克磁珠能吸附500 μg左右的DNA;
l 高通量提取:可同时完成数百个样品的提取;
l 环保无毒:提取方案不含有毒成分。
PCR扩增
PCR,又称聚合酶链式反应,基本原理类似于 DNA 的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。该技术可以从被称为模板DNA的混合物中扩增出一个高数量级特定的目标DNA片段。模板来源有很多种,除了如上述讲到的从细胞提取基因组DNA外,还有从RNA逆转录来的cDNA等。
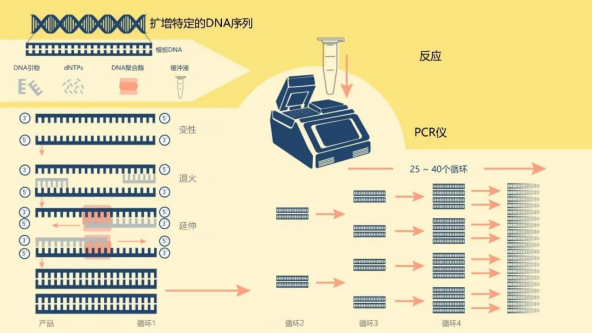
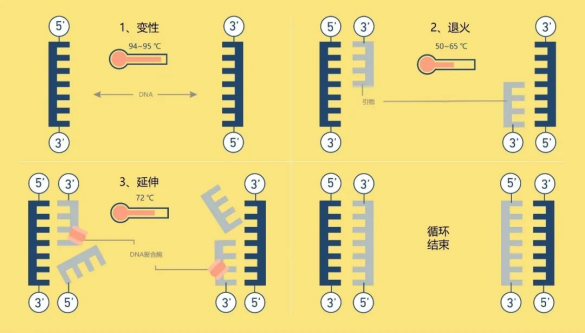
一个PCR反应需要包含以下组分:DNA模板、上下游引物、dNTPs、反应缓冲液、DNA聚合酶等,而通常商业化的试剂会将除DNA模板和上下游引物以外的组分全部预混,以方便科研使用。PCR实验最需要注意的是做好引物设计和反应程序的设置,一般引物设计通过Premier 5或NCBI在线设计等平台来设计。
引物设计注意事项
l 引物 3’端最后一个碱基选择 C 或 G;
l 引物 3’端最后 8 个碱基应避免出现连续错配;
l 引物 3’端尽量避免出现发夹结构;
l 引物 Tm 值控制在 55℃-65℃之间;
l 引物额外附加序列,即与模板非配对序列,不应参与引物 Tm 值计算;
l 引物 GC 含量控制在 40%-60%之间;
l 正向引物和反向引物 Tm 值以及 GC 含量尽可能一致。

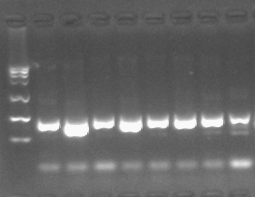
PCR产物鉴定与纯化
PCR产物可通过琼脂糖凝胶电泳技术来鉴定产物的质量,包括得到片段的长度、产量、特异性等。
l 跑胶的目的条带可以通过DNA胶回收试剂盒来回收目标DNA;
l PCR反应产物也可直接通过PCR产物清洁回收试剂盒进行纯化,以用于下游实验。
进一步对于想要获得产物DNA碱基序列详细信息,可将样本送至测序公司进行测序。
PCR优化思路

你送出去设计合成引物的公司会在说明书上提供你的寡核苷酸引物的 Tm,但是我们发现不同的公司根据他们采用的计算方式所提供的 Tm明显不一样。这边推荐IDT website的引物设计工具OligoAnalyzer在优化退火温度方面非常好用,Tm计算公式如下:
Tm= 2(A+T) + 4(G+C)
其中 A, T, G和C表示你的引物序列中对应的碱基数

Tm与缓冲液的盐浓度、pH值以及引物浓度有关,但是一般人很少去考虑这些。大部分人会告诉你,为了防止引物二聚体的形成,推荐你退火温度要比Tm值至少高到10℃。OligoAnalyzer工具在设计引物时也会考虑到模板DNA的二聚体结构。但是我建议退火温度设定值比你两个引物最低的Tm低3℃。例如,如果你的前置引物Tm是62℃,后置引物Tm是61℃,那么设定它们的退火温度为58℃。
如果你拥有一台具有梯度扩增功能的PCR仪,同时具有足够量的模板,你可以进行两到三次的重复试验。我建议设定一定范围的退火温度进行实验,并评估每个温度效果。

在前几循环的PCR反应中设置为较高的退火温度。在每一循环(或两个循环,或三个循环)之后,退火温度Ta降低1~2℃。
那么,具体怎么做呢?使用上面的例子,两条引物的Tm值分别为62℃和61℃,我会设置退火温度Ta在前两个循环是63℃,接下来的两个循环是62℃,然后是61℃、60℃和59℃,共有10循环。然后在58℃温度下进行25个循环。这种技术好处是,前几个循环是非常严格的,所以它是不太可能发生任何非特异性扩增。同时,当退火温度变得更加容易结合引物时,PCR小管就像具有一定量目标扩增子的水库,当水库中已经具有足够量的目标扩增子时,目标扩增子即可在与非特异性序列的引物争夺中取得胜利!
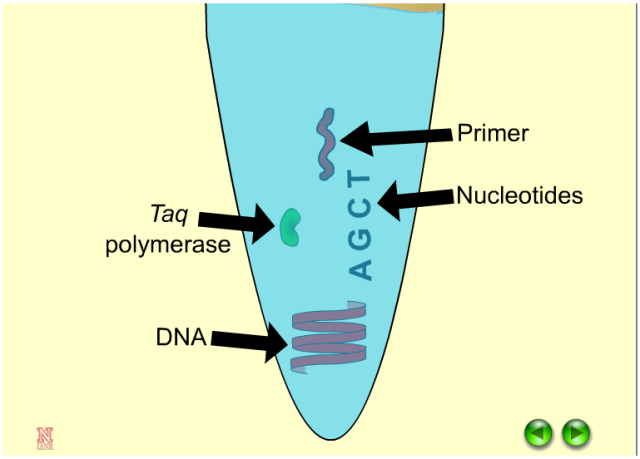
以上实验的前提是确保你的PCR仪能够准确、有效地进行温度调整。我们能理解在一个不能正常工作的PCR仪上会有多少时间被浪费在优化PCR条件,一般来说,你的PCR仪工作不正常,你的PCR反应可能需要比平时花更长的时间。所以在你进行PCR反应时,要确保你的每一步都是能够工作的!包括你的机器、设置的条件以及反应条件、反应成分(即你以前验证过的引物、模板和缓冲条件)。
PCR需要的模板质量会很高!同时,过度的DNA模板含量有可能会降低反应的特异性,增加不必要产物的扩增。引物PCR反应是非常灵敏的,因此尽可能使用较低浓度的模板量。我们推荐的模板量:
质粒DNA模板不超过1 ng;
cDNA:10-40 ng;
基因组DNA:1 μg。

你曾经有没有想过到底是谁想出PCR反应的条件和方法,使得变性和退火时间一直以来都保持一致,只有延伸时间可根据你扩增大小进行调整。一般的经验法则是你每扩增1 kb需要60秒的延伸时间(根据你选用的聚合酶的效率)。例如,如果你扩增一个2 kb的片段,你需设计120秒的延伸时间。对于较短的产物,缩短延伸时间。对于一个200 bp的扩增产物,15到20秒的延伸时间是足够的。延伸时间过长会引入不必要的非特异性的PCR产物!

Taq DNA聚合酶在PCR反应中用的最多,尤其在验证目的基因的PCR扩增中。Pyrococcusfuriosus(PFU)聚合酶则广泛应用于高保真的PCR反应中。

但是也有一些其他的聚合酶对不同的模板有着不一样的扩增效率。比如:如果你的扩增模板中含有较高的GC含量(超过65%),Thermo Scientific的 Accuprime G-C Rich DNA Polymerase扩增的效果将会更好!

当你购买一管DNA聚合酶时,包装里会配备一管已经优化好的Buffer。正常来说,安装说明书进行操作是不会错的。但是我们认为作为一个科研人,了解Buffer组分和浓度对PCR效率的影响很有必要。因为大部分Buffer只是简单地在某个pH值下进行盐离子(KCl和MgCl2)的混合。

可以调整镁离子浓度来改变PCR的效率和保真度。对Taq DNA polymerase,优化的镁离子浓度是1.5到2 mM,因为其他的缓冲液和反应所需的组分有可能会对镁离子产生螯合作用,从而需要增加镁离子浓度。最终在设定镁离子浓度时,会作一个小调整(大约增加0.5 mM)

包括引物长度、GC含量、Tm值、二聚体等等……在这里简单地介绍优化引物浓度。如果添加过多的引物,会增加非特异性结合和引物二聚体。我们认为引物浓度推荐设定为1μM的较低水平效果会好很多。为了提高特异性,引物浓度甚至可以降到0.1 μM,但是可能会减低PCR产量。

dNTPs的浓度不仅会影响PCR的特异性也会影响产量。 太高浓度的dNTP将会降低特异性,太低有可能降低产量。
免责声明:文章内容仅供参考,不构成投资建议。本公众号发布的各类文章重在分享,如有侵权请联系我们,我们将会删除。



推荐新闻
-
- 如何看待qPCR中的那些曲线?
- 2024-04-09深入了解
-
- 如何抓住qPCR数据分析的关键?
- 2024-04-02深入了解
-

- Western Blot实验流程之蛋白样品制备
- 2023-12-26深入了解
-
- 红斑狼疮到底是什么病?每次复发都是重创!
- 2023-12-19深入了解
-

- 我国首个宫颈癌筛查专家共识发布!主推HPV核酸初筛!
- 2023-10-20深入了解

